
- T клеточный лейкоз из был
- Т-клеточная лейкемия/лимфома взрослых
- Т-клеточные лимфомы кожи — грибовидный микоз, синдром Сезари
- Как вылечить Т-клеточный лейкоз
- Что это такое
- Причины
- Неблагоприятный радиационный фон
- Химические соединения
- Вирусные возбудители
- Генетические патологии
- Симптомы
- Стойкое малокровие
- Геморрагический синдром
- Гиперпластические нарушение внутренних органов
- Болевой синдром
- Появление лейкемид
- Диагностирование
- Чем отличается лимфома ходжкина от неходжкинской
- Лечение
- Осложнения
- Прогноз выживаемости при лимфогранулематозе
- Выживаемость
- Профилактика Т-клеточного лейкоза
- T клеточный лейкоз из был
T клеточный лейкоз из был
Т-клеточный крупногранулярный лимфоцитарный лейкоз встречается в 30-50 раз реже В-клеточного ХЛЛ и развивается в возрасте 50-55 лет, чаще у женщин. Основной морфологический признак заболевания — наличие больших гранулярных (содержащих азурофильные гранулы) лимфоцитов в периферической крови и костном мозге. Критерий диагностики — обнаружение больше 2 • 109/л больших гранулярных лимфоцитов в периферической крови. Наиболее частый иммунофенотип: CD3+, CD8+, CD4-, TCRab+.
Лейкемические клетки экспрессируют маркеры апоптоза (Fas или CD95 и Fas-лиганд), но резистентны к Fas-индуцируемому апоптозу.
Спленомегалия выявляется у 20% пациентов, лимфаденопатия и гепа-томегалия встречаются еще реже. В связи с выраженной нейтропенией нередко возникают рецидивирующие инфекции. У 30% больных наблюдается ассоциация с аутоиммунными заболеваниями (аутоиммунная гемолитическая анемия, ревматоидный артрит и др.).
Течение заболевания вариабельно, стандартное лечение не разработано.
Т-клеточная лейкемия/лимфома взрослых
Т-клеточная лейкемия/лимфома взрослых — редкое лимфопролиферативное заболевание, встречающееся преимущественно в странах бассейна Карибского моря и Японии. Доказанным этиологическим фактором является ретровирус HTLV-1. В эндемичных районах инфицировано 5% населения; в течение жизни заболевает один человек из 50-100 инфицированных (в Японии при наличии около 1 миллиона вирусоносителей в год регистрируется около 500 случаев заболевания). Спорадические случаи Т-клеточной лейкемии/лимфомы взрослых отмечены в Европе и Северной Америке.
Опухолевые клетки полиморфны, с полисегментированными, похожими на лепестки цветка, ядрами. Иммунофенотип клеток: CD7-, CD2+, CD3+, CD4+, CD5+, CD25+. Наиболее частые цитогенетические находки — трисомия 12, del 6q.
В подавляющем большинстве случаев Т-клеточная лейкемия/лимфома взрослых характеризуется агрессивным течением, сопровождающимся анемией, лимфаденопатией, гиперкальциемией, ранней диссеминацией (поражение костей, кожи, центральной нервной системы), выраженным иммунодефицитом (обычно дисфункцией CD4+) с развитием тяжелых оппортунистических инфекций. В этих случаях прогноз неблагоприятный (медиана выживаемости не превышает 6 месяцев). Значительно реже встречаются более благоприятные варианты («тлеющий» и хронический), которые, однако, могут трансформироваться в агрессивный тип.
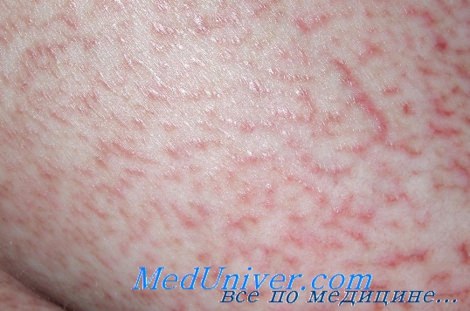 Грибовидный микоз
Грибовидный микоз
Т-клеточные лимфомы кожи — грибовидный микоз, синдром Сезари
Т-клеточные лимфомы кожи — гетерогенная группа лимфопролиферативных заболеваний с первичным поражением кожи. В последние десятилетия заболеваемость Т-клеточными лимфомами кожи достоверно увеличилась. В большинстве случаев встречаются грибовидный микоз и синдром Сезари. Реже наблюдаются первичные СD30-позитивные Т-клеточные лимфопролиферативные заболевания: первичная кожная анапластическая крупноклеточная лимфома (КМ+) и лимфоматоидный папулез.
Грибовидный микоз многие годы протекает с изолированным поражением кожи (эритема, папулы, бляшки, эритродермия), однако в дальнейшем в большинстве случаев развивается агрессивная Т-клеточная лимфома с поражением лимфатических узлов, селезенки, печени и других органов и неблагоприятным исходом. При этом в крови и костном мозге часто обнаруживаются клетки Сезари (перекрестный синдром).
Синдром Сезари — генерализованная Т-клеточная лимфома, характеризующаяся поражением кожи, лимфоаденопатией и наличием в периферической крови неопластически трансформированных Т-лимфоцитов — клеток Сезари (атипичные лимфоциты с большим ядром неправильной формы и скудной базофильной цитоплазмой).
Синдром Сезари характеризуется сходным с грибовидным микозом поражением кожи, однако заболевание сопровождается ранней генерализацией и неблагоприятным течением (лишь 20% больных живут более 5 лет).
Диагноз лимфом кожи доказывается при морфологическом исследовании и результатами иммунофенотипирования (особенно важно это при CD30-позитивных лимфомах). При грибовидном микозе и синдроме Сезари нередко выявляются различные нарушения кариотипа (прежде всего при прогрессировании заболевания). При молекулярно-генетическом исследовании у большинства больных Т-клеточными лимфомами кожи обнаруживается клональная реаранжировка генов Т-клеточных рецепторов.
При локализованных формах Т-клеточных лимфом кожи применяется местное лечение: PUVA-терапия (ультрафиолетовое облучение с фотосенсибилизатором), локальное облучение и введение цитостатиков в опухолевые очаги. При синдроме Сезари используются монохимиотерапия и полихимиотерапия, однако частота ремиссий невелика и они обычно непродолжительны.
источник
Как вылечить Т-клеточный лейкоз


Диагностирование «лейкоза» говорит о том, что в крови человека было выявлено большое количество клеток крови под названием «лейкоциты». Лейкоз бывает острым (в этом случае терапевтическое воздействие должно быть начато уже в первые сутки после выявления заболевания) и хроническим (отличающимся медленным прогрессированием). Т-клеточный лейкоз чаще всего принимает форму хронического заболевания крови.
Содержание
Что это такое
Т-клеточный лейкоз известен под названием лимфома. Он представляет собой раковую опухоль, состоящую из лимфоцитов категории CD-4. Чаще всего она развивается по причине патогенного воздействия вируса Т-клеточного лейкоза.
При отсутствии надлежащего лечения прогрессирование болезни ведёт к нарушениям работы органов, систем и кожных покровов и разрушению суставов и костей. Возможно появление гиперкальциемии. Во время диагностирования в крови пациента обнаруживаются атипично изменённые лимфоциты.
Наиболее высокий порог распространённости отмечается в южной части Японии, на территории островов Карибского бассейна, на берегах Тихого океана и среди жителей Африканского материка. Мужская половина человечества подвержена к развитию данного типа лейкоза больше, чем женщины. Особенно подвержены заражению наркоманы и иные лица, ведущие асоциальный образ жизни.
Причины
Т-клеточный лейкоз развивается при наличии сразу нескольких факторов.
Неблагоприятный радиационный фон
Роль радиационного излучения в провоцировании возникновения рака крови подтверждается на основании выявления внезапного скачка заболеваемости лейкозом среди жителей Японии, после ядерного поражения городов Хиросима и Нагасаки.
Также имеются сведения о повышении числа больных с Т-клеточным лейкозом среди людей, получавших ранее курс радиотерапии в целях борьбы с имеющимися у них онкологическими новообразованиями.
Химические соединения
Наиболее ярко выраженным онкогенным воздействием обладают нефтепродукты. Эти вещества широко используются в индустриальных целях. Соединения бензола и иных нефтепродуктов легко попадают внутрь человеческого тела во время дыхания и сквозь кожные покровы.
Это химическое вещество скапливается в нервных и жировых тканях. Другими химическими соединениями, влияние которых способно приводить к формированию лейкемии, являются пестициды, содержащие галогены, моющие средства и лакокрасочная продукция.
Также установлено, что причиной развития лейкемии могут стать химиотерапевтические фармацевтические вещества, применяющиеся в медицине в целях борьбы с лимфогранулематозом.
Вирусные возбудители
Вирус Т-клеточного лейкоза имеет в себе гены, способные при соприкосновении со здоровыми лейкоцитами крови превращать их в раковые.
Генетические патологии
Лейкозы тесно связаны с наличием таких генетических патологий, как врожденные расстройства деятельности иммунной системы. Поэтому немалую значимость в развитии Т-клеточного лейкоза играет наследственный фактор.
Симптомы
У подавляющего большинства лиц с лейкозом на начальной стадии развития комбинация симптомов может отличаться. Количество и интенсивность симптомов в каждом конкретном случае будет зависеть от состояния иммунной системы и уровня общей физической подготовки человека.
И всё же, в большинстве случаев люди, страдающие Т-клеточным лейкозом, отмечают у себя один или несколько из симптомов.
Стойкое малокровие
Это состояние вызывается подавлением процесса выработки эритроцитов. Она сочетается со стойким незначительным повышением температуры тела, постоянной подавленностью и быстрой утомляемостью, регулярными приступами общего недомогания, потерей сознания и предобморочными состояниями.
Геморрагический синдром
Малое количество зрелых тромбоцитов становится причиной кровоизлияний на кожных покровах и слизистых оболочках, которые могут принимать форму как мелких вкраплений, так и обширных участков. Запущенный геморрагический синдром может стать причиной необъяснимых синяков на коже и сильных кровотечений.
Гиперпластические нарушение внутренних органов
Атипично изменённые лейкоциты приводят к увеличению размеров лимфатических узлов, а также печени и селезёнки.
Болевой синдром
Продолжительная интоксикация организма становится причиной постоянных болей в суставах и костях. Из-за расстройства функционирования иммунной системы больные лейкемией чаще обычного страдают вирусными, инфекционными и бактериальными заболеваниями, что также усугубляет проявления болевого синдрома.
Появление лейкемид
Признаком запущенной стадии лейкоза принято считать появление на поверхности кожных покровов лейкемид – твердых коричневых либо красноватых пятен.
Диагностирование
При начальной стадии течения болезни зачастую раковых клеток в крови ещё очень мало. Чтобы выявить болезнь на ранней стадии, необходимо расширенное исследование клеток гранулярных лейкоцитов при помощи специальных микроскопов.
Если результат исследования выявит, что количество гранулярных клеток существенно превышает установленную норму, то для подтверждения диагноза больному будет назначена проточная цитометрия крови.
Если же раковых клеток ещё совсем мало, пациенту будет проведено исследование «клональности» лейкоцитов. Каждая здоровая клетка лейкоцитов является точнейшей копией друг друга.
По теме

Чем отличается лимфома ходжкина от неходжкинской
Процент схожести лейкоцитов определяют с помощью исследования Т-клеточного рецептора – молекуле, отличающей отдельный лейкоцит от всей остальной лейкоцитарной массы. Проводят данное исследование в лабораториях молекулярно-генетических исследований.
С целью подтверждения наличия заболевания пациенту могут быть назначены дополнительные исследования. В их числе общий анализ крови, патоморфологичское исследование кожных покровов, биохимический анализ, иммуно-ферментный анализ крови и иммунный блоттинг.
Лечение
В наше время пользуется популярностью такой способ лечения Т-клеточного лейкоза, как химиотерапевтическая терапия, заключающаяся в приёме сильнодействующих фармацевтических препаратов. Эти лекарства должны приниматься курсом в виде таблеток и капсул, назначенных лечащим врачом. При отсутствии результатов лечение пациента будет проводиться в стационаре с помощью внутривенных либо внутримышечных инъекций
Ещё один действенный метод – это лучевая терапия. Данный способ лечения лейкозов включает в себя применение рентгеновских лучей или иных видов радиационного излучения.
При необходимости пациенту может быть проведено трансплантирование стволовых клеток. Данный вид вмешательства предназначается для замены патологически изменённых лейкоцитов крови здоровыми клетками.
По назначению лечащего врача могут применяться различные комбинации нескольких методов лечения лейкоза. В последние годы были получены положительные результаты при одновременном применении зидовудина (внутрь) и интерферона группы «а» (в виде подкожных инъекций).
Осложнения
Анемия – неизменное последствие пациентов, страдающих лейкозом. Данное заболевание формируется потому, что поражение костного мозга постепенно сводит на нет возможности для нормального процесса кроветворения.
Эритроциты формируются из лейкемических нормобластов, лейкоз же нарушает процесс нормального формирования эритроидных клеток – предшественников эритроцитов. Всё это приводит к малоэффективному протеканию гемопоэза, из-за чего эритроциты циркулируют в крови более короткий срок.
Нередко у пациентов, страдающих Т-клеточным лейкозом, отмечается и тромбоцитопения. Причина данного состояния связана с поражением костного мозга и резким снижением активности тромбоцитов, что постепенно приводит к появлению геморрагических проявлений.
По теме

Прогноз выживаемости при лимфогранулематозе
Через определённый период времени из-за неполноценности лейкоцитов у пациентов развивается существенное снижение иммунитета, что приводит к повышению чувствительности перед вирусными, микозными и бактериальными возбудителями.
Геморрагические проявления приводят к увеличению печени и селезёнки. При запущенных стадиях заболевания нередко отмечается инфаркт селезенки, требующей отдельной симптоматической терапии.
В числе наиболее частых осложнений при средних и тяжёлых стадиях лейкозов возможно также появление инфильтраций во внутренних органах и в спинном мозге, что приводит к серьёзным нарушениям функционирования различных органов и систем.
В наиболее запущенных стадиях Т-клеточного лейкоза возможно формирование такого состояния, как гиперпуринемия.
Последующая стадия лейкоза — нефропатия, в наиболее запущенной стадии у пациентов нередко отмечается бластный криз.
Выживаемость
Многие дети до 5 лет с диагнозом Т-клеточный лейкоз при наличии должного лечения живут весь период борьбы с болезнью без каких бы то ни было осложнений. В случае если по завершении лечения заболевание никак не проявляет себя в течении 5 лет и более, ребёнок может считаться выздоровевшим.
При проявлениях рецидивирующего Т-клеточного лейкоза достичь ремиссии может быть несколько сложнее. При повторном возникновении болезни спасти жизнь ребёнку и стабилизировать его состояние здоровья в силах трансплантации спинного мозга, которая помогает вторично победить рак у 65%.
Опыт показывает, что одновременное проведение Т–клеточной иммунотерапии и лучевого лечения не только продлевает жизнь людям, но и в разы уменьшает скорость прогрессирования онкологического заболевания и возможных осложнений.
Кроме того, успех лечения зависит от целого ряда факторов, включая эмоциональное состояние самого больного, а также его близких родственников и друзей. При наличии поддержки и умении сохранять положительный настрой шансов на излечение всегда больше.
Профилактика Т-клеточного лейкоза
Обратиться за врачебной помощью следует в случае выявления одного или нескольких симптомов лейкоза. Для недопущения дальнейшего распространения заболевания крови стоит обследовать всех членов семьи, а также половых партнеров больного. Следует помнить, что лица, страдающие Т-клеточным лейкозом, либо страдавшие им в прошлом, не должны быть в числе доноров.
источник
T клеточный лейкоз из был
В середине 70-х годов XX в. одновременно с открытием иммунологических маркеров В- и Т-лимфоцитов появилась возможность разделения этих клеточных популяций. Соответственно двум типам лимфоцитов были выделены две основные группы лимфопролиферативных заболеваний: В- и Т-клеточные. Первый иммунологический тест, позволивший продемонстрировать Т-клеточную природу лимфоцитов, основывался на способности этих клеток формировать розетки при инкубации с эритроцитами барана (Е-розетки).
Совершенствование лабораторных методов исследования, особенно иммунологических и молекулярно-генетических, позволило более детально охарактеризовать и классифицировать эту группу заболеваний. Т-клеточные лимфопролиферативные заболевания составляют 10—15 % от всех опухолей лимфатической системы и по уровню дифференцировки и созревания могут быть разделены на две группы: тимические и посттимические.
Посттимические опухоли представлены иммунологически зрелыми Т-лимфоцитами, в ядрах которых отсутствует фермент терминальная дезоксинуклеотидилтрансфераза (TdT).
Одной из форм зрелоклеточных Т-клеточных лимфопролиферативных заболеваний является Т-клеточный пролимфоцитарный лейкоз (Т-ПЛЛ), впервые описанный D. Catovsky и соавт. в 1973 г.. Наиболее часто Т-клеточный пролимфоцитарный лейкоз (Т-ПЛЛ) встречается в двух возрастных группах: пожилых (средний возраст 69 лет) и молодых с атаксией-телеангиэктазией (AT). Женщины болеют чаще, чем мужчины, в соотношении 4:1. Т-клеточный пролимфоцитарный лейкоз (Т-ПЛЛ) характеризуется агрессивным течением с медианой выживаемости 7,5 мес.
Заболевание начинается остро или подостро. Больные жалуются на быструю утомляемость, слабость, потливость, снижение массы тела. Одними из первых симптомов могут быть боли в животе, связанные с выраженной спленомегалией, увеличением внутри-брюшных лимфатических узлов, а также гематологические изменения (анемия, тромбоцитопения), обусловленные костно-мозговой недостаточностью и гиперспленизмом. Реже первой манифестацией заболевания является поражение кожи, отличающееся полиморфной картиной, — от кожной сыпи, обычно пятнисто-папулезной, до генерализованной эритродермии.
Органные поражения, например ЦНС и легких, встречаются редко. Менее чем у 5 % больных заболевание начинается бессимптомно, и только в анализе крови обнаруживается медленно нарастающий абсолютный лимфоцитоз. Такие случаи, особенно мелкоклеточный вариант Т-клеточного пролимфоцитарного лейкоза (Т-ПЛЛ), ошибочно диагностируют как хронический лимфолейкоз (ХЛЛ). В противоположность ХЛЛ, течение которого может оставаться стабильным длительное время, Т-клеточный пролимфоцитарный лейкоз (Т-ПЛЛ) прогрессирует в течение нескольких месяцев.
Характерным лабораторным изменением при Т-ПЛЛ является высокий лейкоцитоз, который может достигать 1000 • 10 9 /л.
По данным обследования более 100 пациентов Е. Matures и соавт. представили основные клинико-лабораторные проявления Т-клеточного пролимфоцитарного лейкоза (Т-ПЛЛ): спленомегалия — 73 %; лимфаденопатия — 53 %; гепатомегалия — 40%; поражение кожи — 27 %; лейкоцитоз более 100 • 109/л — 75 %; анемия и тромбоцитопения — 30 %.
Этиологические причины Т-клеточного пролимфоцитарного лейкоза (Т-ПЛЛ) не установлены. В сыворотке крови больных не обнаружены антитела к вирусам HTLV-I/II даже у пациентов из эндемичных регионов. С помощью анализа ДНК не удалось доказать наличие геномной последовательности вируса HTLV-I в опухолевых клетках.
Морфологическим субстратом Т-клеточного пролимфоцитарного лейкоза (Т-ПЛЛ) более чем в 2/3 случаев являются пролимфоциты. Они имеют несколько больший по сравнению с обычным лимфоцитом размер, ядро с конденсированным хроматином, которое часто имеет неровные очертания и нуклеолу. Цитоплазма опухолевых клеток базофильная и не содержит гранул. У 20 % больных Т-ПЛЛ опухолевые клетки меньшего размера, в большинстве из них при световой микроскопии нуклеола видна плохо, однако электронно-микроскопическое исследование позволяет выявить ультраструктурные особенности, присущие пролимфоцитам. Такие случаи относят к мелкоклеточному варианту Т-ПЛЛ.
При иммунологическом фенотипировании пролимфоциты, как правило, имеют иммунофенотип CD2+CD5+ и выраженно экспрессируют антиген CD7. Количество CD7-антигенных детерминант на поверхности опухолевых клеток при Т-клеточном пролимфоцитарном лейкозе (Т-ПЛЛ) значительно больше, чем на нормальных Т-лимфоцитах и лимфоцитах при других посттимических Т-клеточных лимфопролиферативных заболеваниях. В 20 % случаев на мембране пролимфоцитов не экспрессируется CD3, однако этот маркер всегда обнаруживается в цитоплазме клеток. Применительно к экспрессии CD4 и CD8 уникального фенотипа, присущего исключительно Т-ПЛЛ, не существует.
Чаще всего (2/3 случаев) пролимфоциты CD4+CD8-, редко фенотип клеток CD8+CD4- и приблизительно в 25 % определяется коэкспрессия CD4 и CD8.
Цитогенетические исследования, проведенные при Т-клеточном пролимфоцитарном лейкозе (Т-ПЛЛ), позволили выявить аномалии хромосом 14, 8 и 11. Перестройки хромосомы 14 составляют 2/3 всех цитогенетических изменений: инверсия хромосомы 14 — invl4(q11q32), тандемная транслокация между двумя хромосомами t(14; 14). Инверсия хромосомы 14 крайне редко встречается при других зрелоклеточных лимфопролиферативных заболеваниях Т-клеточной природы и считается патогномоничной для Т-клеточного пролимфоцитарного лейкоза. Важно отметить сходство цитогенетических изменений в опухолевых клетках при Т-ПЛЛ и в Т-лимфоцитах больных атаксией-телеангиэктазией. Описанные случаи развития Т-клеточных лейкозов у этих пациентов относятся к Т-ПЛЛ.
Как отмечалось, Т-клеточный пролимфоцитарный лейкоз является заболеванием с агрессивным течением. Больные Т-ПЛЛ обычно резистентны к стандартным схемам лечения, включающим алкилирующие препараты (хлорамбуцил, циклофосфамид). Включение в схему терапии антрациклинов (CHOP) позволяет получить ответ, чаще всего частичный и непродолжительный, только у 1/3 больных. Одним из наиболее активных цитостатических препаратов в лечении Т-клеточного пролимфоцитарного лейкоза является 2-деоксикоформицин (пентостатин). Использование его в дозе 4 мг/м2 еженедельно до достижения максимального эффекта позволяет получить общий ответ в 40 % случаев и только в 12 % случаев достигается полная ремиссия. В последние годы предпринимаются успешные попытки использовать при Т-ПЛЛ анти-CD52 моноклональное антитело (Campath-1H).
Иммунотерапия Campath-1H позволяет получить полную ремиссию более чем у половины пациентов, включая резистентных к деоксикоформицину.
источник